突破性算法将材料的探索空间扩展了几个数量级
加州大学圣地亚哥分校雅各布斯工程学院的纳米工程师开发了一种人工智能算法,可以几乎即时地预测任何材料(无论是现有材料还是新材料)的结构和动态特性。该算法被称为M3GNet,用于开发matterverse.ai,这是一个数据库,包含超过3100万种尚未合成的材料,其特性由机器学习算法预测。Matterverse.ai促进了具有卓越性能的新技术材料的发现。
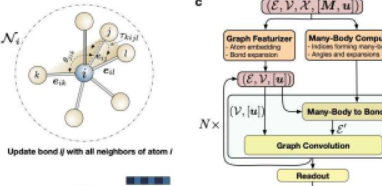
M3GNet背后的团队由加州大学圣地亚哥分校纳米工程教授ShyuePingOng领导,他们使用matterverse.ai和M3GNet的新功能来寻找更安全、能量密度更高的可充电锂离子电池电极和电解质。该项目在11月28日出版的《自然计算科学》杂志上进行了探讨。
材料的性质由其原子排列决定。然而,现有的获得这种安排的方法要么过于昂贵,要么对许多元素无效。
“与蛋白质类似,我们需要了解材料的结构才能预测其特性。”雅各布斯工程学院可持续能源与能源中心副主任Ong说。“我们需要的是用于材料的AlphaFold。”
AlphaFold是谷歌DeepMind开发的一种预测蛋白质结构的人工智能算法。为了构建材料的等价物,Ong和他的团队将图神经网络与多体交互相结合,构建了一个深度学习架构,该架构可以在元素周期表的所有元素中通用、高精度地工作。
“数学图实际上是原子集合的自然表示,”Ong实验室前高级项目科学家、该作品的第一作者ChiChen说,他现在是MicrosoftQuantum的高级量子架构师。“使用图表,我们可以表示材料的全部复杂性,而不会受到传统形式主义中术语组合爆炸的影响。”
为了训练他们的模型,该团队使用了过去十年在材料项目中收集的巨大的材料能量、力和应力数据库。结果是M3GNet原子间势(IAP),它可以预测任何原子集合中的能量和力。Matterverse.ai是通过对无机晶体结构数据库(ICSD)中的5,000多个结构原型进行组合元素替换而生成的。然后使用M3GNetIAP获得平衡晶体结构——一个称为“弛豫”的过程——用于属性预测。
在今天matterverse.ai的3100万种材料中,预计有超过100万种材料具有潜在的稳定性。Ong和他的团队不仅打算大大扩展材料的数量,还打算大幅扩展ML预测属性的数量,包括使用他们之前开发的多保真度方法的小数据量的高价值属性。
除了结构松弛,M3GNetIAP在材料动态模拟和性能预测方面也有广泛的应用。
“例如,我们通常对锂离子在锂离子电池电极或电解质中的扩散速度很感兴趣。扩散越快,电池充电或放电的速度就越快,”Ong说。“我们已经证明,M3GNetIAP可用于以高精度预测材料的锂电导率。我们坚信M3GNet架构是一种变革性工具,可以极大地扩展我们探索新材料化学和结构的能力。”
为了推广M3GNet的使用,该团队已将该框架作为开源Python代码发布在Github上。自2022年2月在Arxiv上发布预印本以来,该团队引起了学术研究人员和业内人士的兴趣。有计划将M3GNetIAP作为工具集成到商业材料模拟包中。
这项工作由加州大学圣地亚哥分校的ChiChen和ShyuePingOng撰写。
免责声明:本文由用户上传,与本网站立场无关。财经信息仅供读者参考,并不构成投资建议。投资者据此操作,风险自担。 如有侵权请联系删除!
-
作为A股市场中极具代表性的黄金珠宝行业龙头企业,作为中国历史最悠久的珠宝品牌之一,老凤祥(股票代码:600612)...浏览全文>>
-
宝子们,杭州 房子装修完成啦!这次要给大家分享几家设计超赞的装修公司哦。它们各具特色,从空间规划到风格...浏览全文>>
-
欲筑室者,先治其基。在上海,装修房子对于每个业主而言,都是极为关键的一步,然而,如何挑选一家值得信赖的...浏览全文>>
-
2025年以来,联通支付严格贯彻落实国家战略部署,以数字和科技为驱动,做好金融五篇大文章,履行支付为民社会...浏览全文>>
-
良工巧匠,方能筑就华居;精雕细琢,方可打造美家。当我们谈论装修公司时,选择一家靠谱可靠的公司是至关重要...浏览全文>>
-
在当今社会,随着城市化进程的高速推进,建筑垃圾的产生量与日俱增。据权威数据显示,我国每年建筑垃圾产生量超 ...浏览全文>>
-
家人们,在上海要装修,选对公司那可太重要了!古语有云:"安得广厦千万间,大庇天下寒士俱欢颜。"一个温馨的...浏览全文>>
-
近年来,新能源汽车市场发展迅猛,各大品牌纷纷推出各具特色的车型以满足消费者多样化的需求。作为国内新能源...浏览全文>>
-
近年来,随着汽车市场的不断变化和消费者需求的升级,安徽滁州地区的宝来2025新款车型在市场上引起了广泛关注...浏览全文>>
-
随着汽车市场的不断变化,滁州地区的消费者对高尔夫车型的关注度持续上升。作为大众品牌旗下的经典车型,高尔...浏览全文>>
- 安徽滁州途安L新车报价2022款,最低售价16.68万起,入手正当时
- 小鹏G7试驾,新手必知的详细步骤
- 别克GL8预约试驾,4S店的贴心服务与流程
- 安徽阜阳ID.4 CROZZ落地价全解,买车必看的省钱秘籍
- 淮北探岳多少钱 2025款落地价,最低售价17.69万起现在该入手吗?
- 安徽淮南大众CC新款价格2025款多少钱能落地?
- 淮北长安启源C798价格,最低售价12.98万起现在该入手吗?
- 安徽淮南途锐价格,各配置车型售价全解析
- 蒙迪欧试驾预约,4S店体验全攻略
- 沃尔沃XC40试驾需要注意什么
- 滁州ID.4 X新车报价2025款,各车型售价大公开,性价比爆棚
- 试驾思域,快速操作,轻松体验驾驶乐趣
- 试驾长安CS35PLUS,一键搞定,开启豪华驾驶之旅
- 天津滨海ID.6 X落地价限时特惠,最低售价25.9888万起,错过不再有
- 天津滨海凌渡多少钱?看完这篇购车攻略再做决定
- 安徽池州长安猎手K50落地价,买车前的全方位指南
- 山东济南ID.6 CROZZ 2024新款价格,最低售价19.59万起,现车充足
- 试驾海狮05EV,新手必知的详细步骤
- 生活家PHEV多少钱 2025款落地价走势,近一个月最低售价63.98万起,性价比凸显
- 奇瑞风云A9试驾,新手必知的详细步骤